|
| ||||
htseq is a python package that provides an infrastructure to process and analyze high-throughput sequencing data. here, we will use htseq to count the number of aligned sequence reads that overlap with a list of genomic features.
to get started, first download and install htseq (prerequisites = { x : python > 2.4 < 3.0, pysam, numpy, matplotlib } :
before running htseq, it is important to note that it includes three overlap resolution modes that dictate how aligned reads that overlap more than one genomic feature are treated. the three overlap resolutions modes are { union, intersection-strict, intersection-nonempty }. see figure 1 for an illustration on the effect on counting of these three modes. for a full list of htseq counting options, consult the man page.
now we are ready to use htseq to generate a list of counts per genomic feature. in this example, we will generate a list of counts per gene.
by default, htseq outputs the count table to stdout, which is the text terminal that initiated the command. to redirect the output to a file called 'aligned.htseq.counts', use bash's redirection operator:
to get started, first download and install htseq (prerequisites = { x : python > 2.4 < 3.0, pysam, numpy, matplotlib } :
wget --no-check-certificate https://pypi.python.org/packages/source/H/HTSeq/HTSeq-0.6.1.tar.gznext, leave the htseq install directory, and download the aligned sequence reads and the genomic annotation set provided on this blog post. the data is a subset of the data found in the pasilla bioconductor package.
tar -xzf HTSeq-0.6.1.tar.gz
cd HTSeq-0.6.1
python setup.py install --user
wget http://www.weebly.com/uploads/2/6/8/5/26850053/aligned.bamthe aligned sequence reads are stored in a binary sequencing alignment/map format, and the genomic annotation set is in the gtf format.
wget http://www.weebly.com/uploads/2/6/8/5/26850053/chr4.gtf.gz
before running htseq, it is important to note that it includes three overlap resolution modes that dictate how aligned reads that overlap more than one genomic feature are treated. the three overlap resolutions modes are { union, intersection-strict, intersection-nonempty }. see figure 1 for an illustration on the effect on counting of these three modes. for a full list of htseq counting options, consult the man page.
now we are ready to use htseq to generate a list of counts per genomic feature. in this example, we will generate a list of counts per gene.
python -m HTSeq.scripts.count --format=bam --minaqual=0 --stranded=no --type=exon --idattr=gene_name --mode=union aligned.bam chr4.gtf.gzthe command tells htseq to only consider exons when counting, and to sum up all the counts by the gene name. the output is a tab delimited table of read counts for each gene listed in the genomic annotation set, followed by a series of special counters, which count reads which were not counted for a variety of reasons, including { no_feature, ambiguous, too_low_aQual, not_aligned, alignment_not_unique }.
by default, htseq outputs the count table to stdout, which is the text terminal that initiated the command. to redirect the output to a file called 'aligned.htseq.counts', use bash's redirection operator:
python -m HTSeq.scripts.count --format=bam --minaqual=0 --stranded=no --type=exon --idattr=gene_name --mode=union aligned.bam chr4.gtf.gz > aligned.htseq.countsusing the head command, you can examine the first five lines of the output file:
head -n 5 aligned.htseq.counts
# ATPsyn-beta 0
# Actbeta 0
# Ank 6750
# Arf102F 0
# Asator 205
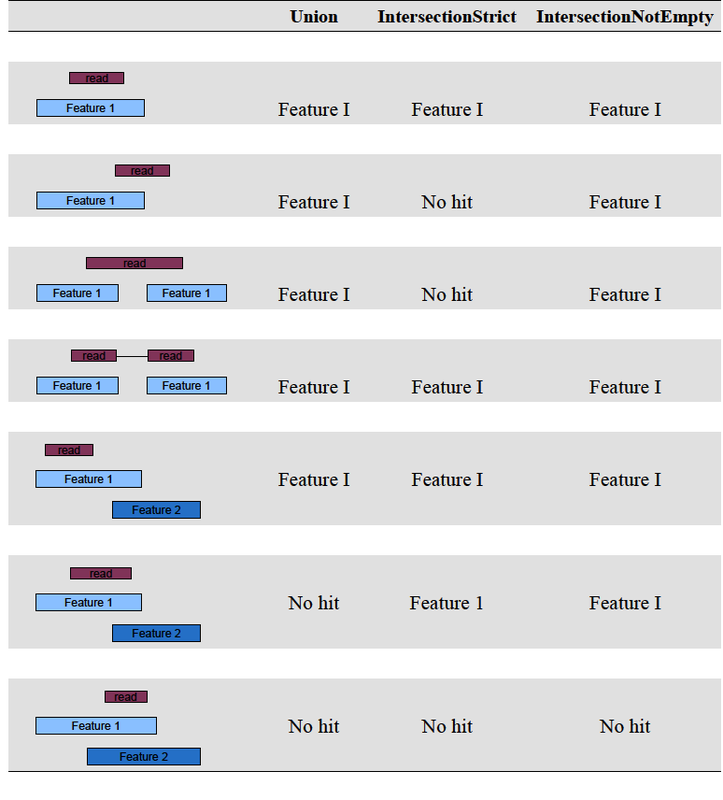
figure 1: the three overlap modes of htseq. *image from genomic-ranges bioconductor package: http://www.bioconductor.org/packages/release/bioc/html/GenomicRanges.html
 RSS Feed
RSS Feed